Department of Chemistry, Faculty of Science/Graduate School of Science/School of Science, Kyushu University
Research Introduction
Development of a Quantitative pKa Prediction Method Using Electronic Structure Theory for Molecules in Solution
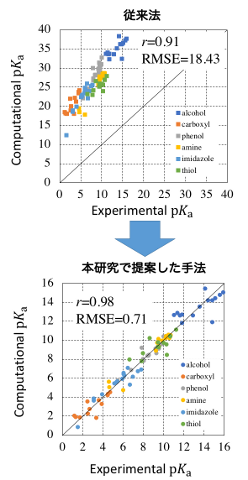 The acid dissociation constant (pKa) is a value that defines the proton dissociation tendency of a molecule and is one of the important indicators in the analysis of biopolymers such as proteins and DNA, whose structure and properties change depending on the presence of hydrogen bonds. This value can be obtained experimentally by nuclear magnetic resonance (NMR) or neutron diffraction (ND). However, the former method is difficult to assign proton peaks, while the latter method requires large crystals for analysis and is difficult to produce. Therefore, in recent years, studies to predict this value theoretically have been attracting attention. The strength of the theoretical approach is that it does not require real crystals, and the pKa value can be obtained only by calculation. However, it is difficult in practice to provide a quantitative value because the state of protons in aqueous solution may not be strictly known.
The acid dissociation constant (pKa) is a value that defines the proton dissociation tendency of a molecule and is one of the important indicators in the analysis of biopolymers such as proteins and DNA, whose structure and properties change depending on the presence of hydrogen bonds. This value can be obtained experimentally by nuclear magnetic resonance (NMR) or neutron diffraction (ND). However, the former method is difficult to assign proton peaks, while the latter method requires large crystals for analysis and is difficult to produce. Therefore, in recent years, studies to predict this value theoretically have been attracting attention. The strength of the theoretical approach is that it does not require real crystals, and the pKa value can be obtained only by calculation. However, it is difficult in practice to provide a quantitative value because the state of protons in aqueous solution may not be strictly known.
Therefore, in this study, we developed a method for quantitative pKa prediction by an approach using linearity, based on a theoretical method for calculating pKa values based on thermodynamic cycles. Based on the approach for pKa prediction by linear fitting using the PCM method by Matsui et al. at the University of Tsukuba, this study avoids the rigorous treatment of proton states and ensures quantitativity. In addition, in this study, solvent effects are incorporated using the 3D-RISM method to reflect more rigorous intermolecular interactions and achieve improved calculation accuracy.
Glycan Recognition Mechanism of Glycoconjugate Binding Modules and Ion Species Dependence at the Binding Site
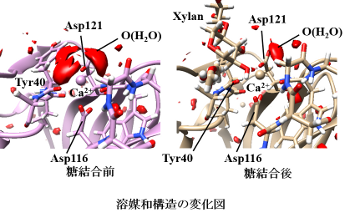
One of the most important chemical processes that occurs in living organisms is protein molecular recognition. This is the selective and specific binding of proteins to certain ligands, of which enzyme substrate recognition is a typical example. It is not only the direct interaction between protein and ligand that characterizes the selectivity of molecular recognition. Interactions with the myriad of water molecules around them play an equally important role.
In our laboratory, we use two approaches to theoretical analysis of protein molecular recognition: molecular dynamics simulation and 3D-RISM theory. Molecular dynamics simulations are used to analyze the structure, arrangement, and interaction of proteins and ligands, while 3D-RISM theory is used to analyze the distribution and interaction of water molecules. 3D-RISM theory is one of the integral equation theories of liquids, and it is capable of treating a myriad of water molecules based on statistical mechanics. In this study, we used this method to elucidate the molecular mechanism of carbohydrate (polysaccharide) recognition of a protein called carbohydrate-binding module family 36 (CBM36), especially the role of metal ions in the binding site in the recognition of carbohydrate chains.
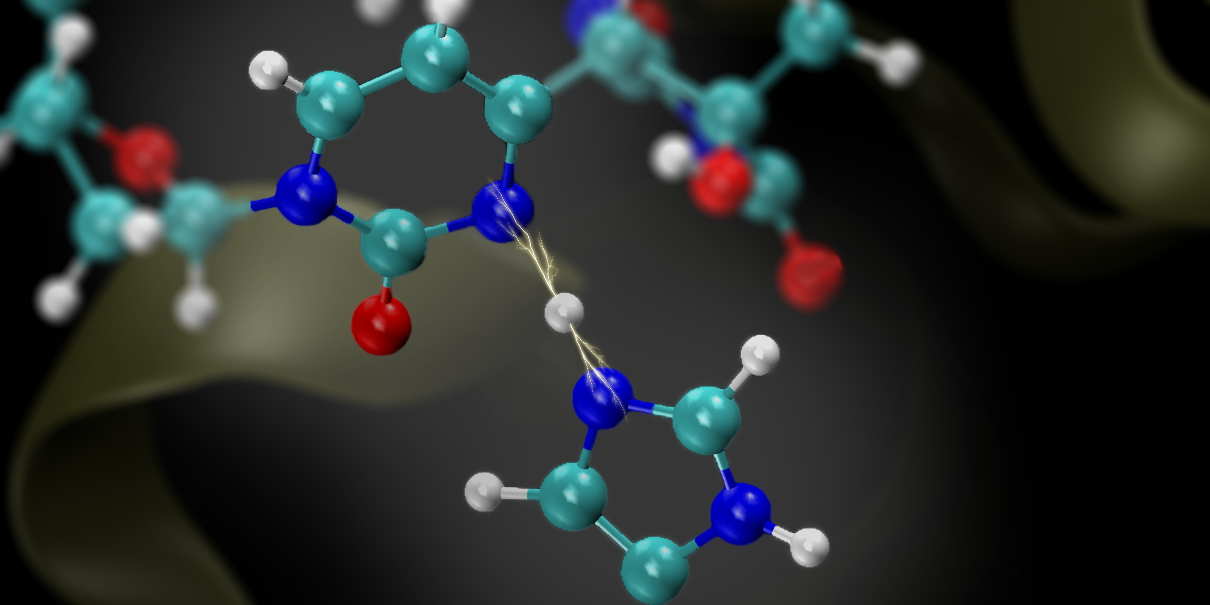
Proton transfer reaction of glycine in mixed solvents
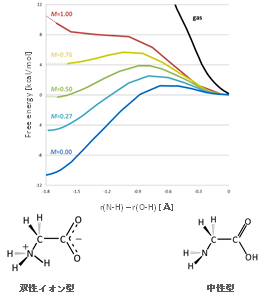
Proton transfer reactions are one of the fundamental chemical reactions and are known to be strongly affected by solvents. One such reaction is the intramolecular proton transfer of amino acids. Since amino acids have amino and carboxyl groups, they take a zwitterionic conformation in polar solvents such as water and a neutral conformation in less polar solvents. However, there are no detailed reports on the structure of these amino acids in mixed solvents or their proton transfer reaction profiles.
In this study, we performed a theoretical study to understand intramolecular proton transfer reaction profiles and solvent effects in aqueous and acetonitrile mixtures using glycine as an example, calculated with the RISM-SCF method, which is applicable to solute-solvent systems. The RISM-SCF method allows us to incorporate molecular interactions of solvent molecules, such as hydrogen bonding, and to treat mixtures of solvents of arbitrary mixing ratios in a non-empirical manner.
Development of a new relativistic molecular orbital method
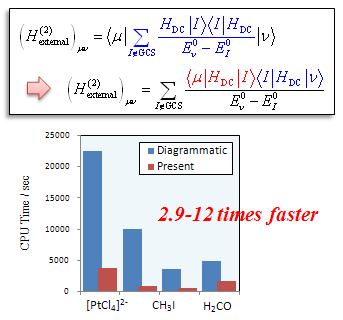
Molecules containing heavy molecules such as lanthanides are widely used as materials for light-emitting devices. In order to control the wavelength and luminance, it is useful to obtain a detailed electronic structure by high-precision calculations, but it is necessary to incorporate relativistic effects when dealing with such systems.
The computational cost required to account for relativistic effects can be very high, limiting the size of applicable systems. Therefore, the development of algorithms to improve computational efficiency can reduce the computational cost while maintaining the accuracy of the calculation. This will make it possible to compute larger systems. In this study, we proposed the relativistic GMC-QDPT, which is a highly accurate calculation method, and made it possible to calculate large systems such as transition metal complexes.
Aromaticity and excitation spectra of phosphorus-containing porphyrins
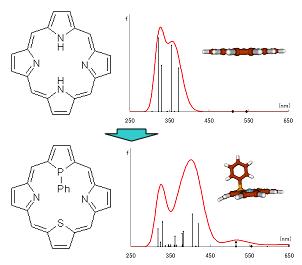
Porphyrins are macrocyclic aromatic compounds with abundant absorption and emission properties and are the basic backbone of molecules that play important roles in biological systems. Because of its properties, porphyrins are used in a variety of applications, including molecular devices and functional materials. For this reason, many studies have been conducted both theoretically and experimentally, and control of their physical properties is expected. In this study, we evaluated the effect of phosphorus substitution on the structure, aromaticity, and excitation spectra of phosphorus-containing porphyrins by theoretical calculations.
In particular, we will clarify the planar structure and high aromaticity of free base porphyrins, how the 18π-conjugated system is changed by phosphorus substitution, and the excitation spectral changes and the factors behind these differences.
Excited states of coumarin derivatives
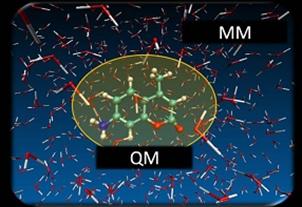
Actual chemical reactions occur not only in the gas phase but also in solvents. Therefore, when performing theoretical calculations for systems in which the presence of a solvent is considered to have a significant effect, it is essential to incorporate the effect of the solvent. However, it is difficult to perform calculations that take into account all the electrons in the solution from the viewpoint of computational cost, and efficient solvent modeling is necessary. In this study, we used the QM/MM MD method as a solvent model to study solvent effects on the electron spectrum of C120, one of the 7-aminocoumarin derivatives.
Radicality of the ground state of acenes
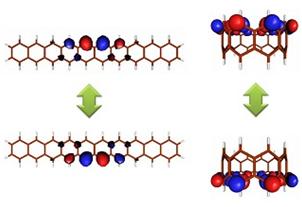
Acenes are fused benzene rings, among which oligoacenes and cyclasenes are expected to be used as organic semiconductors and memory devices. They can be regarded as a part of carbon nanotubes, and it is important to gain knowledge of their electronic states for future development. In this study, we focused on the diradicality of the ground state and performed systematic calculations even for molecules with a large number of rings, which cannot be synthesized experimentally, and evaluated their radicality.
